CPSI Deficiency: A Comprehensive Guide
CPSI deficiency, or carbamoyl phosphate synthetase I deficiency, is a rare but serious genetic disorder that affects the urea cycle. This cycle is crucial for removing ammonia, a toxic waste product, from the body. When CPSI, the first enzyme in this cycle, is deficient, ammonia builds up in the blood, leading to hyperammonemia. This condition can cause severe neurological damage and even death if left untreated. This comprehensive guide provides an in-depth look at CPSI deficiency, covering its causes, symptoms, diagnosis, and management. We aim to provide you with the most current and comprehensive information available, drawing upon expert consensus and the latest research to empower you with knowledge and understanding of this complex condition.
What is CPSI Deficiency?
CPSI deficiency is an inherited metabolic disorder. It disrupts the normal function of the urea cycle, which is responsible for converting ammonia into urea, a less toxic substance that can be excreted in urine. The deficiency arises from mutations in the CPS1 gene, which provides instructions for making the carbamoyl phosphate synthetase I enzyme. This enzyme is essential for initiating the urea cycle within the mitochondria of liver cells.
When CPSI is deficient, the urea cycle cannot effectively process ammonia. This leads to a buildup of ammonia in the blood, a condition known as hyperammonemia. Hyperammonemia is particularly dangerous because ammonia is toxic to the brain. Elevated ammonia levels can cause neurological damage, leading to developmental delays, seizures, coma, and even death. The severity of CPSI deficiency can vary widely, depending on the specific mutation in the CPS1 gene and the amount of functional CPSI enzyme that is produced. Some individuals may experience symptoms early in infancy, while others may not develop symptoms until later in childhood or even adulthood.
Understanding the nuances of CPSI deficiency is crucial for early diagnosis and effective management. While rare, recognizing the signs and symptoms and initiating prompt treatment can significantly improve outcomes for affected individuals.
Genetic Basis and Inheritance of CPSI Deficiency
CPSI deficiency is caused by mutations in the CPS1 gene, located on chromosome 2. This gene provides the instructions for producing the carbamoyl phosphate synthetase I enzyme. Different mutations in the CPS1 gene can lead to varying degrees of enzyme deficiency, influencing the severity of the condition.
The inheritance pattern of CPSI deficiency is autosomal recessive. This means that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition. Individuals who inherit only one copy of the mutated gene are carriers. Carriers typically do not show symptoms of CPSI deficiency because they have one functional copy of the CPS1 gene that produces enough CPSI enzyme to maintain normal urea cycle function. However, carriers can pass the mutated gene to their children.
If both parents are carriers of a CPS1 gene mutation, there is a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene and develop CPSI deficiency, a 50% chance that their child will inherit one copy of the mutated gene and become a carrier, and a 25% chance that their child will inherit two normal copies of the gene and will not be affected or a carrier.
Symptoms and Diagnosis of CPSI Deficiency
The symptoms of CPSI deficiency can vary depending on the severity of the enzyme deficiency and the age of onset. In newborns, symptoms typically appear within the first few days of life. These may include:
- Lethargy
- Poor feeding
- Vomiting
- Irritability
- Seizures
- Respiratory distress
- Coma
In older children and adults, symptoms may be milder and more intermittent. They can include:
- Recurrent vomiting
- Ataxia (lack of coordination)
- Slurred speech
- Behavioral changes
- Intellectual disability
- Seizures
- Episodes of hyperammonemia triggered by illness or stress
Diagnosing CPSI deficiency involves a combination of clinical evaluation and laboratory testing. Suspicion of CPSI deficiency often arises based on the patient’s symptoms, particularly if they have a history of unexplained hyperammonemia. Diagnostic tests typically include:
- Plasma ammonia levels: Elevated ammonia levels in the blood are a key indicator of urea cycle disorders, including CPSI deficiency.
- Plasma amino acid analysis: This test can help identify characteristic patterns of amino acid abnormalities associated with CPSI deficiency.
- Urine organic acid analysis: This test can help rule out other metabolic disorders that may cause similar symptoms.
- Enzyme assay: A liver biopsy can be performed to measure the activity of CPSI enzyme directly. This is the most definitive diagnostic test for CPSI deficiency.
- Genetic testing: Genetic testing of the CPS1 gene can identify specific mutations that cause CPSI deficiency. This can confirm the diagnosis and provide information about the severity of the condition.
Management and Treatment of CPSI Deficiency
The primary goal of treatment for CPSI deficiency is to lower ammonia levels in the blood and prevent hyperammonemic episodes. Treatment strategies typically involve a combination of dietary management, medications, and, in some cases, liver transplantation.
Dietary Management
Dietary management is a cornerstone of treatment for CPSI deficiency. The goal is to restrict protein intake to minimize ammonia production while ensuring adequate nutrition for growth and development. Dietary strategies include:
- Protein restriction: The amount of protein allowed in the diet is carefully calculated based on the patient’s age, weight, and tolerance.
- Specialized formulas: Infants with CPSI deficiency are often fed specialized formulas that are low in protein and supplemented with essential amino acids.
- Frequent feedings: Frequent feedings help to maintain stable blood glucose levels and prevent catabolism, which can lead to increased ammonia production.
- Supplementation: Carnitine supplementation may be used to help remove excess acyl-CoA, which can inhibit the urea cycle.
Medications
Several medications are used to help lower ammonia levels in patients with CPSI deficiency. These include:
- Sodium benzoate and sodium phenylacetate: These medications help to remove ammonia from the blood by providing alternative pathways for nitrogen excretion. They react with glycine and glutamine, respectively, to form compounds that are excreted in the urine.
- Ammonium phenylbutyrate (Buphenyl): This medication is a prodrug that is converted to phenylacetate in the body. Phenylacetate then reacts with glutamine to form phenylacetylglutamine, which is excreted in the urine.
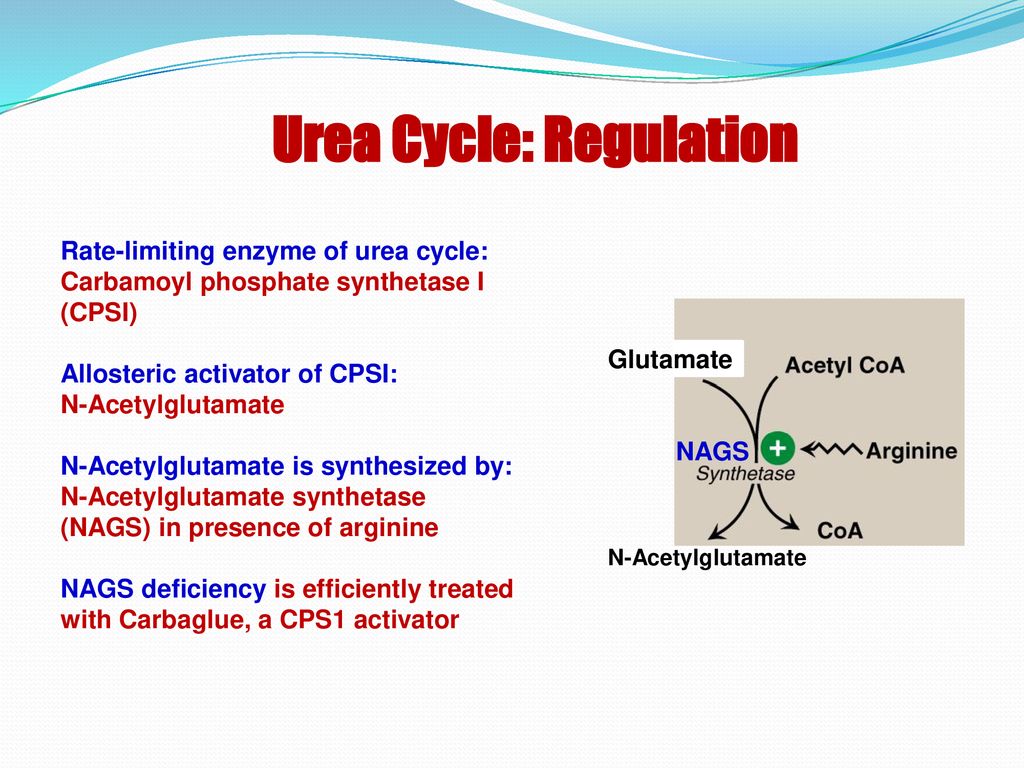
- N-carbamylglutamate (Carbaglu): This medication is a synthetic analog of N-acetylglutamate (NAG), which is an essential activator of CPSI. Carbaglu can help to improve the activity of residual CPSI enzyme in patients with partial CPSI deficiency.
Liver Transplantation
Liver transplantation is a curative treatment option for CPSI deficiency. A transplanted liver provides a source of functional CPSI enzyme, restoring normal urea cycle function. Liver transplantation is typically considered for patients with severe CPSI deficiency who do not respond adequately to dietary management and medications. While liver transplantation can significantly improve outcomes, it is a major surgical procedure with potential risks and complications. Patients who undergo liver transplantation require lifelong immunosuppression to prevent rejection of the transplanted organ.
Emergency Management of Hyperammonemic Crises
Hyperammonemic crises are life-threatening complications of CPSI deficiency. Prompt recognition and treatment are essential to prevent neurological damage and death. The management of hyperammonemic crises typically involves:
- Stopping protein intake: All protein intake should be stopped immediately to reduce ammonia production.
- Administering intravenous fluids: Intravenous fluids help to dilute the ammonia in the blood and promote its excretion in the urine.
- Administering medications: Medications such as sodium benzoate, sodium phenylacetate, and arginine are used to lower ammonia levels.
- Hemodialysis: Hemodialysis may be necessary to remove ammonia from the blood quickly in severe cases of hyperammonemia.
- Monitoring: Close monitoring of ammonia levels, electrolytes, and neurological status is essential.
The Role of Genetic Counseling
Genetic counseling is an important part of the management of CPSI deficiency. Genetic counselors can provide information about the inheritance pattern of CPSI deficiency, the risk of recurrence in future pregnancies, and the availability of genetic testing. Genetic counseling can help families make informed decisions about family planning and prenatal testing.
Prenatal testing is available for CPSI deficiency if the mutations in the CPS1 gene have been identified in the parents. Prenatal testing options include chorionic villus sampling (CVS) and amniocentesis. Preimplantation genetic diagnosis (PGD) is also an option for couples undergoing in vitro fertilization.
Living with CPSI Deficiency
Living with CPSI deficiency requires ongoing management and monitoring. Patients and their families need to work closely with a team of healthcare professionals, including metabolic specialists, dietitians, and genetic counselors. Adherence to dietary restrictions and medication regimens is essential to prevent hyperammonemic episodes and maintain good health.
Support groups and online communities can provide valuable resources and support for individuals and families affected by CPSI deficiency. Connecting with others who understand the challenges of living with this condition can help to reduce feelings of isolation and provide practical advice and emotional support. Our experts emphasize the importance of continued research and advocacy to improve the lives of those affected by CPSI deficiency.
Understanding the Impact of CPSI Deficiency on Development
CPSI deficiency, if not properly managed, can significantly impact a child’s development. Hyperammonemia, the hallmark of the condition, is particularly damaging to the developing brain. Early and consistent management is crucial to minimize these developmental impacts.
Neurological consequences can range from mild learning disabilities to severe intellectual disability, depending on the frequency and severity of hyperammonemic episodes. Motor skills can also be affected, leading to delays in achieving developmental milestones such as sitting, crawling, and walking. In some cases, CPSI deficiency can lead to seizures, which can further impact neurological development.
However, with early diagnosis and consistent adherence to treatment protocols, many children with CPSI deficiency can achieve near-normal developmental outcomes. Regular monitoring of ammonia levels, meticulous dietary management, and appropriate medication are key to mitigating the risk of neurological damage. A multidisciplinary approach involving metabolic specialists, dietitians, neurologists, and developmental therapists is essential to optimize developmental outcomes for children with CPSI deficiency.
Future Directions in CPSI Deficiency Research
Research into CPSI deficiency is ongoing, with the goal of developing new and improved treatments. Areas of active research include:
- Gene therapy: Gene therapy aims to correct the underlying genetic defect that causes CPSI deficiency by delivering a functional copy of the CPS1 gene to liver cells.
- Enzyme replacement therapy: Enzyme replacement therapy involves administering a purified form of the CPSI enzyme to patients with CPSI deficiency.
- Novel medications: Researchers are working to develop new medications that can lower ammonia levels more effectively and with fewer side effects.
Continued research and innovation are essential to improving the lives of individuals and families affected by CPSI deficiency. As our understanding of the condition grows, we can expect to see the development of more effective treatments and improved outcomes.
Seeking Expert Guidance and Support
Navigating the complexities of CPSI deficiency requires a collaborative approach with experienced healthcare professionals. Early diagnosis, meticulous management, and ongoing support are crucial for optimizing outcomes and improving the quality of life for individuals affected by this rare condition. Remember, you are not alone. A dedicated team of experts, including metabolic specialists, dietitians, genetic counselors, and support groups, are available to provide guidance, resources, and emotional support every step of the way. By working together, we can empower individuals with CPSI deficiency to live full and meaningful lives.
